Scientists at St. Jude Children’s Research Hospital explored how mutations in mitochondrial DNA contribute to cancer, the extent of their impact, and when and how they become a factor. Mitochondria act as energy factories in cells and have their own, separate DNA. Mutations to mitochondrial DNA (mtDNA) have been observed in cancer, but it has been unclear how these changes might affect cancer growth. To find answers, St. Jude Children’s Research Hospital scientists combined computational tools and DNA sequencing technologies to examine these mtDNA mutations in cancer cells closely. Their new method lets scientists pinpoint when these mutations occur, how they change as cancer develops and whether they affect how cancer cells behave. The results of this study were published today in Science Advances.
Exploring the role that individual mtDNA mutations have on cancer has historically been difficult. “Each cell contains hundreds of copies of mitochondrial DNA; so, a mutation might be present at low levels in many cells, or at high levels in just a subset of cells,” said corresponding author Mondira Kundu, MD, PhD, St. Jude Department of Cell & Molecular Biology. “These different patterns can have dramatically different effects on how cells function.”
mtDNA mutations are not random passengers in cancer
To overcome this challenge, the team combined several techniques, including powerful computational tools, statistical analyses, bulk whole genome sequencing and single-cell studies. This approach allowed them to determine how much mitochondrial DNA was mutated in each cell, and when these changes happened in relation to cancer development. Surprisingly, the researchers found that some mitochondrial DNA mutations occur before a cell turns cancerous — and that these mutations are not always random. It appears that in some cases, cancer cells actively “select” for a mix of normal and mutated mitochondrial DNA.
“This approach allowed us to tell apart harmless ‘passenger’ mutations from those that may help cancer grow,” Kundu explained. “That’s something the field has struggled with until now.”
Kundu’s team took the analysis further by deploying a tool, called NetBID2, created by co-author Jiyang Yu, PhD, St. Jude Department of Computational Biology interim chair. With this tool, the researchers found evidence that mtDNA may contribute to therapy resistance. They discovered a mtDNA mutation linked to changes in pathways associated with resistance to glucocorticoids, a common therapy for acute lymphoblastic leukemia. Further analysis suggested that this type of mitochondrial mutation may make leukemia cells more likely to resist treatment.
While this research highlights the role mitochondrial DNA mutations might play in leukemia, the main achievement is the creation of a novel multidimensional approach to investigate mtDNA. Kundu is optimistic about the value of digging deeper into this overlooked feature of cancer growth.
“This work shows that mitochondrial DNA can influence both how leukemia starts and how it progresses,” said Kundu. “The next important step is to apply this approach to many more patient samples, so we can fully understand its impact.”
We’re told we get our mitochondria from our moms, and that’s true.
In humans, mitochondrial DNA is almost exclusively maternally inherited. Sperm mitochondria do enter the egg, but they’re typically tagged for destruction—marked and cleared by mitophagy so the embryo keeps mom’s mitochondrial line.
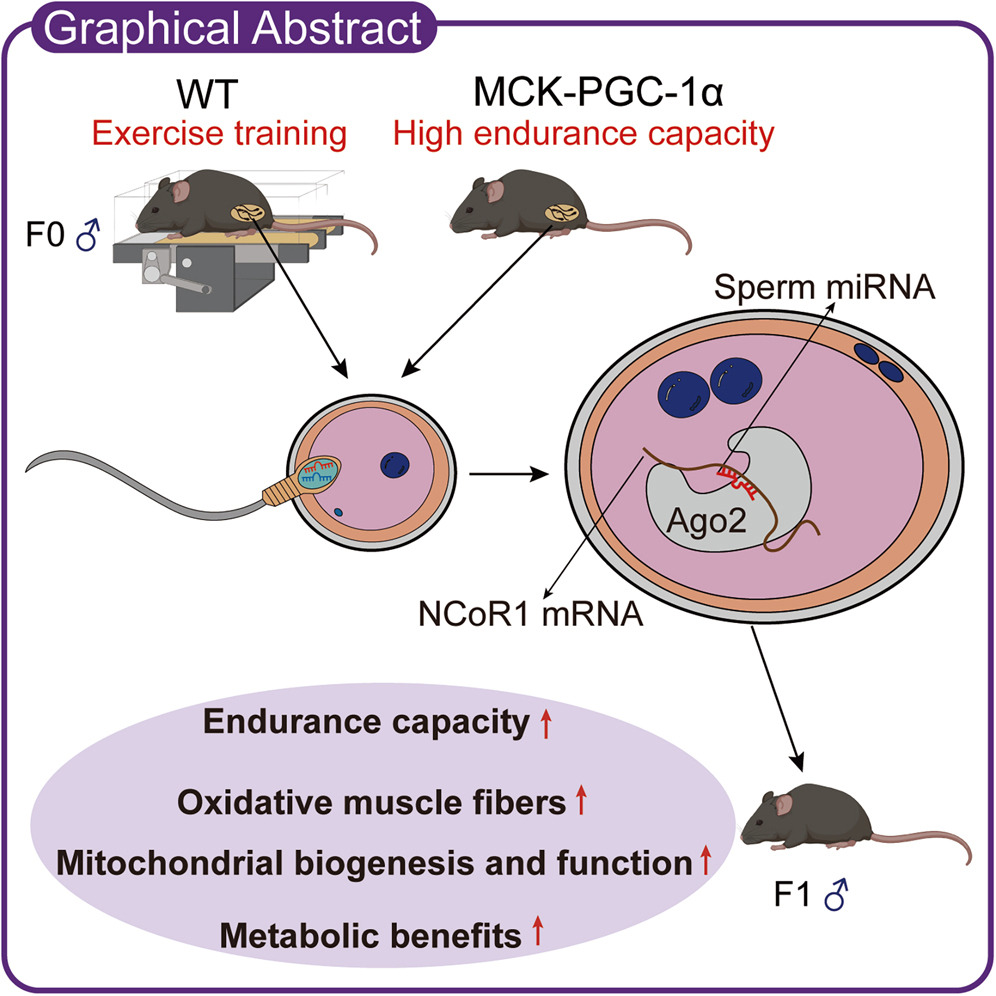
But even though mitochondrial DNA comes from mom, dad’s workouts before conception may still shape a child’s endurance capacity and metabolic health, not by passing on his mitochondria, but by sending tiny sperm microRNA messages that tune early embryonic gene regulation.
In a new study, exercising fathers produced offspring with greater endurance and more mitochondria, an effect that tracked to sperm small RNAs that suppress a “molecular brake” on PGC-1α, the classic mitochondrial biogenesis switch.
The findings reframe preconception health as a two-parent story—mom supplies the mitochondria, but dad’s training status can still program how those mitochondria are used.
Untrained offspring inherit their father’s fitness
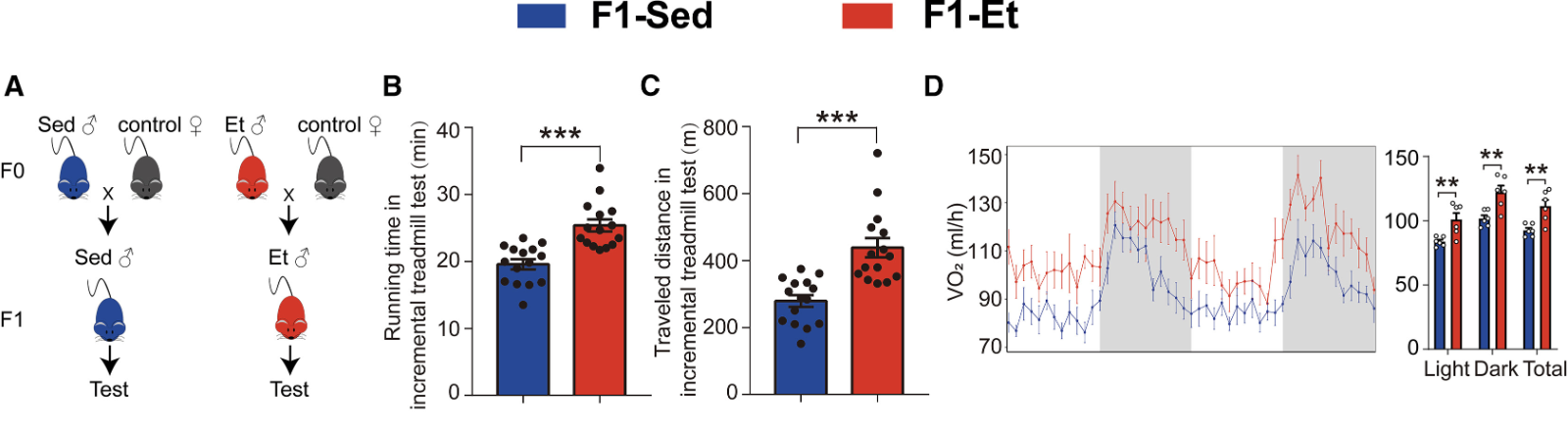
For the study, male mice completed 8 weeks of progressive endurance exercise training on a treadmill, a program that successfully enhanced their fitness. Compared to their untrained peers, they were leaner, had higher bone mineral density, superior endurance, greater energy expenditure, increased mitochondrial abundance, more fatigue-resistance type I muscle fibers, and (at the molecular level), increased expression of PGC-1α—the master metabolic regulator.
This better fitness and endurance showed up in their offspring. When male mice trained before conception, their offspring (who never trained) ran longer and farther (with lower post-exercise lactate levels) than mice from sedentary fathers, and their leg muscles looked like those of trained mice, with more slow-twitch/oxidative fibers, higher mitochondrial enzyme activity, and visibly more mitochondria.
They also mirrored their fathers’ body composition, with higher lean mass, lower fat mass, and improved bone mineral density compared to offspring of sedentary fathers. Metabolically, endurance-trained offspring exhibited higher oxygen consumption and burned more calories throughout the day.
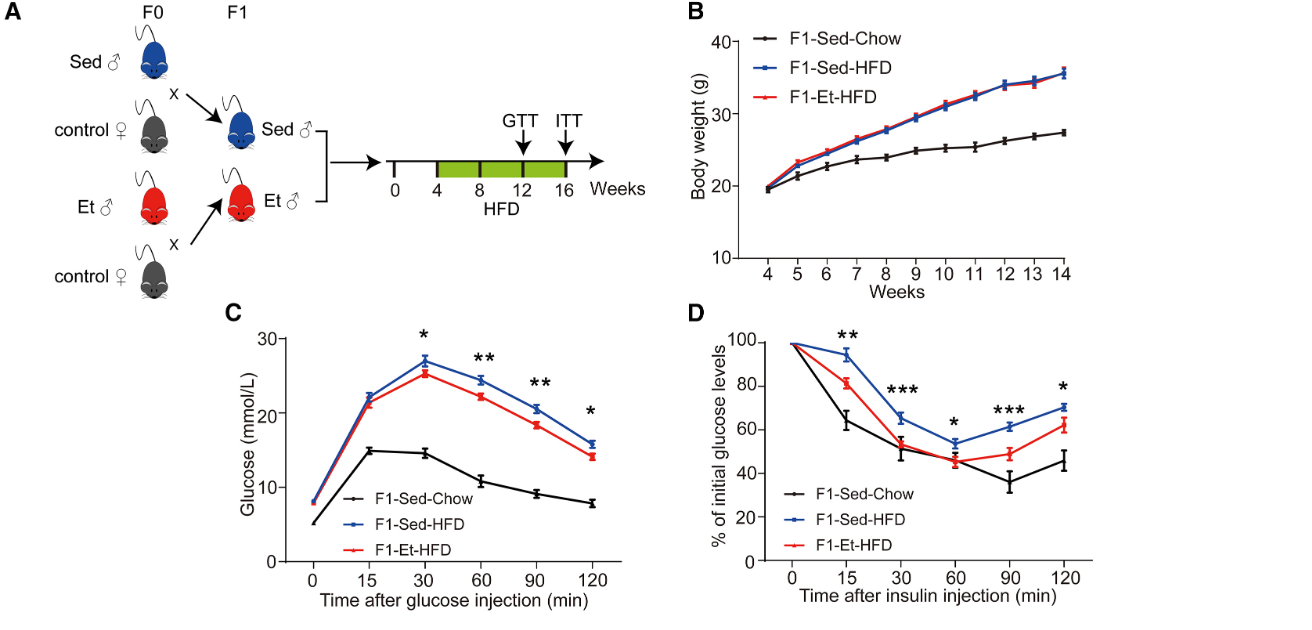
Put on a high-fat diet, offspring of exercising fathers had better blood sugar control than those of sedentary fathers. Skeletal muscle was pinpointed as the main driver. Their muscles pulled in more glucose, stored more glycogen, showed stronger insulin signaling, and had more of the glucose transporter GLUT4.
Endurance capacity (B and C) and energy expenditure (D) in offspring of sedentary (blue) and endurance-trained (red) male mice.
PGC-1α’s crucial role
A complementary experiment showed the same effect without any treadmill training.
Fathers engineered to overexpress PGC-1α—the muscle’s “fitness switch”—passed on endurance benefits even when the transgene wasn’t inherited. Offspring that did inherit the transgene had a 31% higher peak VO₂ than controls, and even the wild-type littermates (whose fathers overexpressed PGC-1α but who lacked the gene themselves) showed a 14% increase in peak VO₂. Even when pups did not inherit the genetic tweak, they still showed the endurance and mitochondrial advantages, set up through a non-genetic mechanism.
Small sperm RNAs carry the message
Researchers isolated RNA from the sperm of exercising fathers and injected it into normal embryos. This alone recreated the full package of fitness and metabolic benefits in otherwise standard offspring, who had a leaner body composition, better endurance, and more mitochondria in their muscle.
The tiny RNAs worked by dialing down an embryonic brake called NCoR1, a corepressor that acts as a brake on PGC-1α-driven mitochondrial biogenesis. Exercise and elevated PGC-1α in fathers produced a shared signature of altered sperm microRNAs, several of which target NCoR1.
Injecting just one of those microRNAs (miR-148a-3p) into standard embryos reduced embryonic NCoR1 and was enough to produce adult mice with higher endurance and more oxidative muscle.
Body weight (B) and glucose tolerance (C and D) in offspring from sedentary (blue) and endurance-trained (red) male mice fed a high-fat diet.
Fitness-forward genes are conserved in humansTo test whether this pathway shows up in people, the researchers compared trained and untrained men. The trained group had markedly higher aerobic capacity (VO₂max of ~63 vs. ~53), confirming a meaningful endurance phenotype before looking at sperm biology. They then profiled the same exercise-responsive sperm microRNAs highlighted in the mouse experiments. Of the ten miRNAs consistently elevated after paternal exercise or PGC-1α activation in mice, seven are conserved in humans and all of them were significantly higher in sperm from the trained men. Is that amount of microRNA enough to matter at fertilization? Maybe. The study estimated ~830 vs. ~2,779 copies per sperm in sedentary vs. exercised males. Prior work suggests ~100 copies per cell can repress targets—so these amounts are plausibly active during the earliest embryonic stages. In short: endurance training is associated with the same conserved sperm-miRNA pattern in humans and exercised mice, making it biologically plausible that a father’s training status before conception could influence early embryonic gene regulation (even though child outcomes weren’t measured in this study).
DOI: 10.1016/j.cmet.2025.09.003
The importance of parental exercise
Unsurprisingly, most of the research on parental habits and childhood health has focused on the role of the mother before and during pregnancy—her body weight, what she eats and drinks, and other lifestyle habits she engages in or avoids. That’s especially true when it comes to exercise. The role of the father, however, is a bit more hazy.
For moms, randomized controlled studies show that exercise during pregnancy reduces the risk of pregnancy complications (macrosomia, abnormal vaginal delivery, C-section) and lowers the odds of gestational diabetes, hypertension, and preeclampsia—which pose short- and long-term health risks for mother and newborn.[1][2] There’s also possibly autonomic and neurodevelopmental advantages, for example, better language and cognitive development in children at age 2 and 5 if their mothers exercised regularly.[3]
For dads, human evidence is limited to molecular-level studies such as the one discussed today—we don’t have much information on long-term outcomes after paternal exercise, even though the mechanistic plausibility is there.
But we do know that fitness can be passed down. VO2 max trainability (how much one can improve their fitness via training) is estimated to have 47% heritability, and even endurance performance measures like lactate threshold show significant parent-offspring resemblance—highly trainable and fit parents are likely to have fitter, highly trainable kids.[4][5] Part of this is environmental, but part is also genetic. It’s not just fitness either—parents (particularly fathers) who are highly active have children who are more likely to be highly physically active throughout childhood and into adulthood. While this might reflect modeled lifestyle habits rather than inheritance per se, there’s likely a genetic component at play too.
Many chronic diseases can be traced to mitochondrial dysfunction, according to Chen Junxu, a natural medicine expert at Bastyr University. After reviewing over 500 research papers and drawing from his extensive clinical practice, Chen developed a comprehensive theory about the relationship between mitochondrial health and chronic disease, which he shared in a recent interview on NTDTV’s “Health 1+1“ program.
Understanding Mitochondria’s Vital Role
Mitochondria are often called the power generators of human cells. They convert nutrients such as glucose and fatty acids that we obtain from food into adenosine triphosphate (ATP), the primary energy source in our cells during metabolism.
At the same time, mitochondria are the core of human immunity, too. Healthy mitochondria effectively regulate immune responses, while mitochondrial dysfunction can damage immune cells, resulting in many chronic diseases and impaired cellular differentiation.
Chen argues that seemingly diverse conditions—including diabetes, hypertension, heart disease, cancer, allergies, autoimmune diseases such as rheumatoid arthritis, and even various mental illnesses—can be understood through a “unified theory” of mitochondrial imbalance. This means that almost every disease can be traced to mitochondrial imbalance. In other words, in mitochondrial imbalance, there is invariably something wrong with the body’s basic metabolism. This perspective suggests that approximately 90 percent of chronic diseases stem from problems with mitochondrial metabolism.